



People >
Lude Franke
While over 10,000 genetic risk factors are now known for diseases such as diabetes and cancer, it is still unclear through what mechanisms these risk factors eventually cause these diseases. For most of these risk factors it remains unclear what genes they change, in what cell-types or tissues these effects manifest themselves and what pathways get disrupted.
Our group uses gene expression data to investigate this and to get to a better understand-ing how genetic risk factors cause disease. In the last few years we have developed new large-scale ‘big data’ approaches (see e.g. Westra et al, Nature Genetics 2013, Fehrmann et al, Nature Genetics 2015, Pers et al, Nature Communications 2015, Bonder et al, Nature Genet-ics 2017 and Zhernakova et al, Nature Genetics 2017), to use new RNA-seq data but also to ‘recycle’ large amounts of existing gene expression data.
Lude Franke (1980) is associate professor in systems genetics (Department of Genetics, University Medical Centre Groningen). He has over 13 years of research experience on the development and application of statistical and computational methods for performing functional genomics. In particular he has performed several large-scale expression quantita-tive trait locus (eQTL) mapping analysis, showing that disease-associated SNPs typically have regulatory effects.
He studied biomedical sciences (Utrecht University) where he obtained his PhD in 2008 (cum laude) while developing new, cutting-edge computational and statistical methods for conducting genome-wide association studies (GWAS) and gene network reconstruction using gene expression data. After graduation, he took up concurrent postdoc positions in London (Institute of Cell and Molecular Science) and Groningen (University Medical Centre Gronin-gen). In London he conducted research on the genetics of immune-mediated diseases (i.e. celiac disease) and in Groningen he worked on the development of methods to identify the effects of genetic risk factors on gene expression levels (Franke and Jansen, Methods Mol Bio 2009). His post-doc work resulted in a paper (Dubois et al, Nature Genetics 2010) that combined his work in London and Groningen. In this paper he demonstrated that there were many independent genetic risk-variants in celiac disease and that they mostly increase disease-risk by altering gene expression levels.
As senior author, he subsequently developed various computational methods and software to increase statistical power to identify such effects on gene expression (Westra et al, Bioin-formatics 2010, Fehrmann et al, PLoS Genetics 2011, cited >100x). By using these methods, he was able to demonstrate that the genetic risk-variants for many other diseases also have an effect on gene-expression levels, and that these genetic variants often affect gene expres-sion levels in only specific cell types (Fu et al, PLoS Genetics 2012, Westra et al, under review). He showed that SNPs affect the expression levels of many long non-coding genes (lncRNAs, Kumar et al, PLoS Genetics, January 2013) and that they can affect poly-adenylation of genes (Zhernakova et al, PLoS Genetics, June 2013). Through a large-scale blood eQTL meta-anal-ysis consortium that he initiated in 2010, he identified downstream (trans-eQTL) effects for over 100 different risk-SNPs (Westra et al, Nature Genetics 2013). His work has demon-strated that functional genomics approaches enable previously unknown pathways for many different diseases to be identified. He recently showed in a reanalysis of 80,000 gene expres-sion profiles that somatic copy number aberrations in cancer can be detected well when cor-recting such data for ‘transcriptional components’ (Fehrmann et al, Nature Genetics 2015). These transcriptional components also permit accurate prediction of gene functions, used in pathway analysis method DEPICT (Pers et al, Nature Communications 2015) to use these predicted gene functions to better interpret GWAS findings, and developed strategies to integrate genetic variation, gene expression and methylation data. Recent last-author papers include Zhernakova et al, Nature Genetics 2017 and Bonder et al, Nature Genetics 2017.
Currently the lab is concentrating on integrating multi-omics datasets, such as as conducting large-scale trans-QTL meta-analyses in >30,000 samples, in conjunction with single-cell RNA-seq data, to identify likely causal genes for diseases that might be targetable by drugs, and to help develop computational strategies to increase the diagnostic yield in clinical genetics, by integrating RNA-seq, whole-genome sequencing data and gene function prediction.
In 2014 his first PhD student (Harm-Jan Westra) obtained his PhD (cum laude). Dasha Zherna-kova followed in 2016 and Marc Jan Bonder obtained his PhD (cum laude) in 2017. He is cur-rently supervising several PhD students and Postdocs (made possible by an NWO VIDI Grant and ERC Starting Grant).
Biological interpretation of genome-wide association studies using predicted gene functions.
Pers TH, Karjalainen JM, Chan Y, Westra HJ, Wood AR, Yang J, Lui JC, Vedantam S, Gustafsson S, Esko T, Frayling T, Speliotes EK; Genetic Investigation of ANthropometric Traits (GIANT) Consortium, Boehnke M, Raychaudhuri S, Fehrmann RS, Hirschhorn JN, Franke L. Nature Communications 2015 Jan 19;6:5890. (Cited > 100x)
Gene expression analysis identifies global gene dosage sensitivity in cancer. Fehrmann RS, Karjalainen JM, Krajewska M, Westra HJ, Maloney D, Simeonov A, Pers TH, Hirschhorn JN, Jansen RC, Schultes EA, van Haagen HH, de Vries EG, Te Meerman GJ, Wijmenga C, van Vugt MA, Franke L. Nature Genetics 2015 (Cited > 100x)
Identification of context-dependent expression quantitative trait loci in whole blood.
Zhernakova DV, Deelen P, Vermaat M, van Iterson M, van Galen M, Arindrarto W, van ‘t Hof P, Mei H, van Dijk F, Westra HJ, Bonder MJ, van Rooij J, Verkerk M, Jhamai PM, Moed M, Kiel-basa SM, Bot J, Nooren I, Pool R, van Dongen J, Hottenga JJ, Stehouwer CD, van der Kallen CJ, Schalkwijk CG, Zhernakova A, Li Y, Tigchelaar EF, de Klein N, Beekman M, Deelen J, van Heemst D, van den Berg LH, Hofman A, Uitterlinden AG, van Greevenbroek MM, Veldink JH, Boomsma DI, van Duijn CM, Wijmenga C, Slagboom PE, Swertz MA, Isaacs A, van Meurs JB, Jansen R, Heijmans BT, ‘t Hoen PA, Franke L. Nature Genetics 2017 Jan;49(1):139-145
Disease variants alter transcription factor levels and methylation of their binding sites.
Bonder MJ, Luijk R, Zhernakova DV, Moed M, Deelen P, Vermaat M, van Iterson M, van Dijk F, van Galen M, Bot J, Slieker RC, Jhamai PM, Verbiest M, Suchiman HE, Verkerk M, van der Breggen R, van Rooij J, Lakenberg N, Arindrarto W, Kielbasa SM, Jonkers I, van ‘t Hof P, Nooren I, Beekman M, Deelen J, van Heemst D, Zhernakova A, Tigchelaar EF, Swertz MA, Hof-man A, Uitterlinden AG, Pool R, van Dongen J, Hottenga JJ, Stehouwer CD, van der Kallen CJ, Schalkwijk CG, van den Berg LH, van Zwet EW, Mei H, Li Y, Lemire M, Hudson TJ; BIOS Consor-tium, Slagboom PE, Wijmenga C, Veldink JH, van Greevenbroek MM, van Duijn CM, Boomsma DI, Isaacs A, Jansen R, van Meurs JB, ‘t Hoen PA, Franke L, Heijmans BT. Nature Genetics 2017 Jan;49(1):131-138
We offer various research projects and are always looking for talented and motivated people who are enthusiastic and are eager to learn about genetics, bioinformatics, statistical, math-ematics and bioinformatics. You must have completed or are working towards a BsC or MSc degree in (Medical) Biology, Biochemistry, Mathematics, Physics, Bioinformatics or Computer Science or in a closely related field.
You are interested in the following topics:
- Design, develop, implement, and maintain new algorithms, applications and infrastructure components (primarily using Java and occasionally using R)
- Analysis of genomics, proteomics and metabolomics datasets
- Data visualization: Since we are working with ‘big data’ it is very important to think about ways to visualize such data, to better understand what we are working on.
Ideally you should have experience in medical biology (or equivalent), mathematics, physics, bioinformatics or computer science.
We believe it is very important that you are curious and eager to learn new things, and that you have already strong skills in either mathematics, statistics, bioinformatics, programming or data visualization.
Since you will work in an international team with several post-docs, PhD students and techni-cians from the Netherlands, China, Russia, Finland, Poland, India, Mexico and Italy, you need to be able to speak, read and write english well.
Most importantly though: If you feel you can learn from us, please do not hesitate to get in touch by sending an e-mail and your curriculum vitae to lude@ludesign.nl
Synopsis: New method makes larger studies into the origin of cancer possible
Funding:
Cancer arises due to mutations in the DNA. But in order to identify these mutations, it is necessary to study the DNA of very many cancer patients. Researchers at the University Medical Centre Groningen (UMCG) have developed a method to do this and have now analysed the data from 16,000 cancer patients. This is one of the largest oncology studies to date, worldwide.
The new method was developed by a team un-der the leadership of Prof. Lude Franke, a statis-tical geneticist: “The systematic analysis of DNA from 16,000 tumours is very costly. However, in the past 15 years, studies have looked at a lot of gene expression and these measurements are publically available. We have developed a new statistical method so that we can re-assess this information. By investigating more than 16,000 tumours, we have been able to determine which changes occur in the DNA. We saw that certain mutations in the tumour DNA are very common, while others occur only in specific tumour types, like breast cancer.”
In recent years, scientists all over the world have collected large quantities of genetic data, but analysing this ‘big data’ is a huge challenge. An international group of researchers headed by scientists from the University Medical Center Groningen (UMCG) have now succeeded. By ana-lysing large quantities of data on genes and gene expression with new technologies, for about a hundred different diseases they could work out which processes become imbalanced in the body, even before people actually get sick.
The researchers investigated the DNA of 8,000 people and studied the consequences of DNA mutations. ‘We discovered that for many dis-eases the mutations all disrupted a single bio-logical process’, says Dr Lude Franke, the main researcher on the project. This was completely unknown for many of these diseases. Franke gives the example of how insulin-dependent di-abetes (type I diabetes) occurs. ‘We discovered that a mutation is responsible for a certain im-
Medical-oncologist Dr. Rudolf Fehrmann pointed out that this method makes it possible to look at gene expression profiles in a fresh light com-pared to the past 15 years. “It has enabled us to indicate potential starting points for develop-ing new therapies for a group of cancers that are difficult to treat, for example, (a group that has many mutations in the DNA). These are now be-ing investigated with experiments in the labora-tory.”
The researchers studied 80,000 expression pro-files in developing their method. Such a ‘big data’ approach has only recently become possible, with the arrival of better computers and new mathematical techniques that permit very effi-cient research to be performed. Large amounts of data, which were gathered for completely dif-ferent purposes, are now proving useful to fur-ther insight into how cancer arises. This method makes new studies in this field possible and will save much money.
mune reaction becoming too extreme. This then functions as a “trigger” thus disrupting a process and lead-ing to illness.’ Franke hopes that the new insights will contribute to the development of new drugs for diabetes, directed towards inhibiting this im-mune response.
According to Franke, this heralds a new phase in genetic research. ‘Researchers are traditionally inclined to formulate a single specific research question, gather data and then come to a conclu-sion. However, with the arrival of big data, better computers and new mathematical techniques, it is now possible to conduct colossal studies that can provide the answers to a large number of different questions at the same time. This new approach has enabled us to acquire detailed in-sight into what exactly goes wrong in the body for a large number of different diseases, even before someone actually gets sick. This provides starting points for the development of drugs that correct these disrupted biological processes.’
Synopsis: ‘Big data’ revolution provides new insight into the origin of diseases
A complete overview of all publicatons can be found at Google Scholar

We develop computational and statistical methods to better understand how genetic risk factors eventually cause disease. We aim to elucidate which processes go wrong in what cell-types and tissues, by generating new bio-logical datasets and reanalysis existing (public) datasets.
Key publications
News
We feel passionate about
gaining new scientific insight
by reinterpreting large datasets
Vacancies and internships
Key themes:
Data visualization
Key themes:
Statistical modelling





How to get in touch
Franke Lab
Department of Genetics, 5th floor
European Research Institute for the Biology of Ageing (ERIBA)
Antonius Deusinglaan 1
9713 AV Groningen
The Netherlands
Lude@ludesign.nl
The department of Genetics is part of the University Medical Center Groningen (UMCG) and is involved in the research of multiple complex diseases. We have contributed to the identi-fication of genetic variants that cause type 1 diabetes, rheuma-toid arthitis, celiac disease, ulcerative colitis and Crohn’s disease.
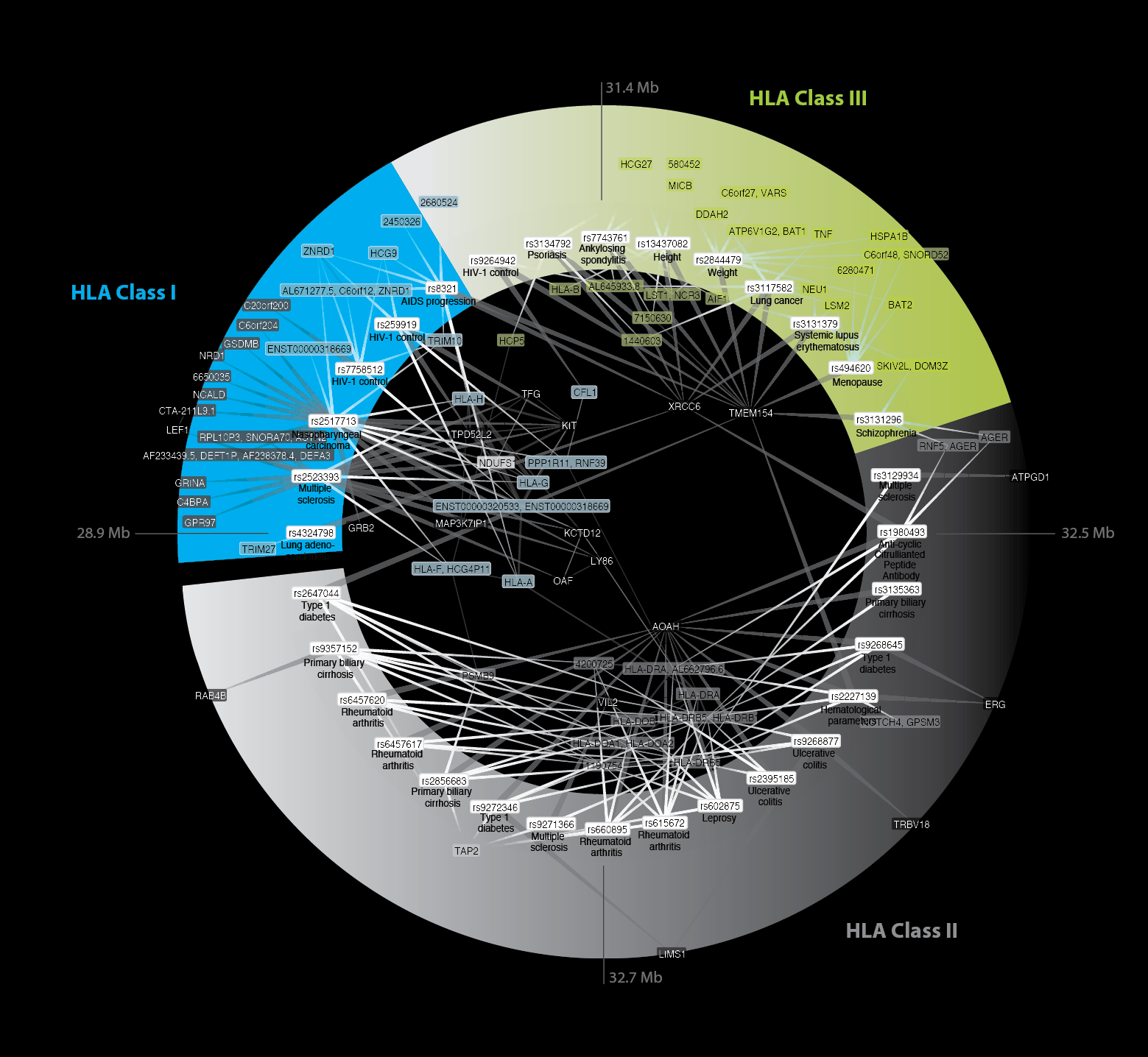
Systematic identification of trans eQTLs as putative drivers of known disease associations. Westra HJ, Peters MJ, Esko T, Yaghootkar H, Schurmann C, Kettunen J, Christiansen MW, Fairfax BP, Schramm K, Powell JE, Zhernakova A, Zhernakova DV, Veldink JH, Van den Berg LH, Karjalainen J, Withoff S, ... Franke L. Systematic identification of trans eQTLs as putative drivers of known disease associations. Nature Genetics 2013 (Cited > 600x)
Human disease-associated genetic variation impacts large intergenic non-coding RNA expres-sion. Kumar V, Westra HJ, Karjalainen J, Zhernakova DV, Esko T, Hrdlickova B, Almeida R, Zher-nakova A, Reinmaa E, Võsa U, Hofker MH, Fehrmann RS, Fu J, Withoff S, Metspalu A, Franke L, Wijmenga C. PLoS Genetics 2013 (Cited > 150x)
Unraveling the regulatory mechanisms underlying tissue-dependent genetic variation of gene expression. Fu J, Wolfs MG, Deelen P, Westra HJ, Fehrmann RS, Te Meerman GJ, Buurman WA, Rensen SS, Groen HJ, Weersma RK, van den Berg LH, Veldink J, Ophoff RA, Snieder H, van Heel D, Jansen RC, Hofker MH, Wijmenga C, Franke L. PLoS Genetics 2012 (Cited > 150x)
Trans-eQTLs reveal that independent genetic variants associated with a complex phenotype converge on intermediate genes, with a major role for the HLA. Fehrmann RS, Jansen RC, Veld-ink JH, Westra HJ, Arends D, Bonder MJ, Fu J, Deelen P, Groen HJ, Smolonska A, Weersma RK, Hofstra RM, Buurman WA, Rensen S, Wolfs MG, Platteel M, Zhernakova A, Elbers CC, Festen EM, Trynka G, Hofker MH, Saris CG, Ophoff RA, van den Berg LH, van Heel DA, Wijmenga C, Te Meerman GJ, Franke L. PLoS Genetics 2011 (Cited > 200x)
MixupMapper: correcting sample mix-ups in genome-wide datasets increases power to detect small genetic effects. Westra HJ, Jansen RC, Fehrmann RS, te Meerman GJ, van Heel D, Wij-menga C, Franke L. Bioinformatics 2011
Multiple common variants for celiac disease influencing immune gene expression. Dubois PC, Trynka G, Franke L, Hunt KA, Romanos J, Curtotti A, Zhernakova A, ... Wijmenga C, van Heel DA. Nature Genetics 2010 (Cited > 600x)



Map of the Netherlands
Visual designed by Lude Franke,
using OpenGL, Java and Adobe Illustrator
Visual designed by Lude Franke using Java2D and iText
while using data mining and principal component analysis
Learn about:
- Genetics
- Java
- Statistics
- R
- Data mining
- Mathematics
- Big data
- Bioinformatics
We often use data mining approaches to learn pat-terns in data that we previously were not aware of.
We often use data mining approaches to learn pat-terns in data that we previously were not aware of.
Key themes:
Big data
Key themes:
Data integration







Key themes:
Data visualization
Visual designed by Lude Franke using Java2D and
iText while using eQTL data and force-directed layout
We integrate public & private datasets to gain new insight
















Groningen City Centre
Franke Lab
Single-cell RNA sequencing identifies celltype-specific cis-eQTLs and co-expression QTLs.
Monique G. P. van der Wijst, Harm Brugge, Dylan H. de Vries, Patrick Deelen, Morris A. Swertz, LifeLines Cohort Study, BIOS Consortium, Lude Franke. Nature Genetics 2018 Apr;50(4):493-497
Harm-Jan obtains
his PhD cum laude
Dasha
obtains
her PhD
Marc Jan obtains
his PhD cum laude
Marc Jan 1st
author on
trans-meQTL
paper in Na-
ture Genetics
Lude obtains VIDI grant
Lude obtains ERC Starting grant
Harm-Jan obtains
Rubicon grant
2015
2016
2017
2014
2013
Harm-Jan 1st author on eQTL paper in Nature Genetics
Dasha 1st au-thor on eQTL paper in PLoS Genetics
Juha 1st author on gene network paper in Nature Genetics
Dasha 1st author
on context-specific
eQTL paper in
Nature Genetics
Marc Jan 1st author on meQTL paper in BMC Medical Genom-ics








2018
Monique 1st author
on single cell
eQTL paper in
Nature Genetics
Juha
obtains
his PhD
